Nothing Happens Suddenly: Looking Beyond Risk to Find Disease Before the Event
Nothing Happens Suddenly: Looking Beyond Risk to Find Disease Before the Event
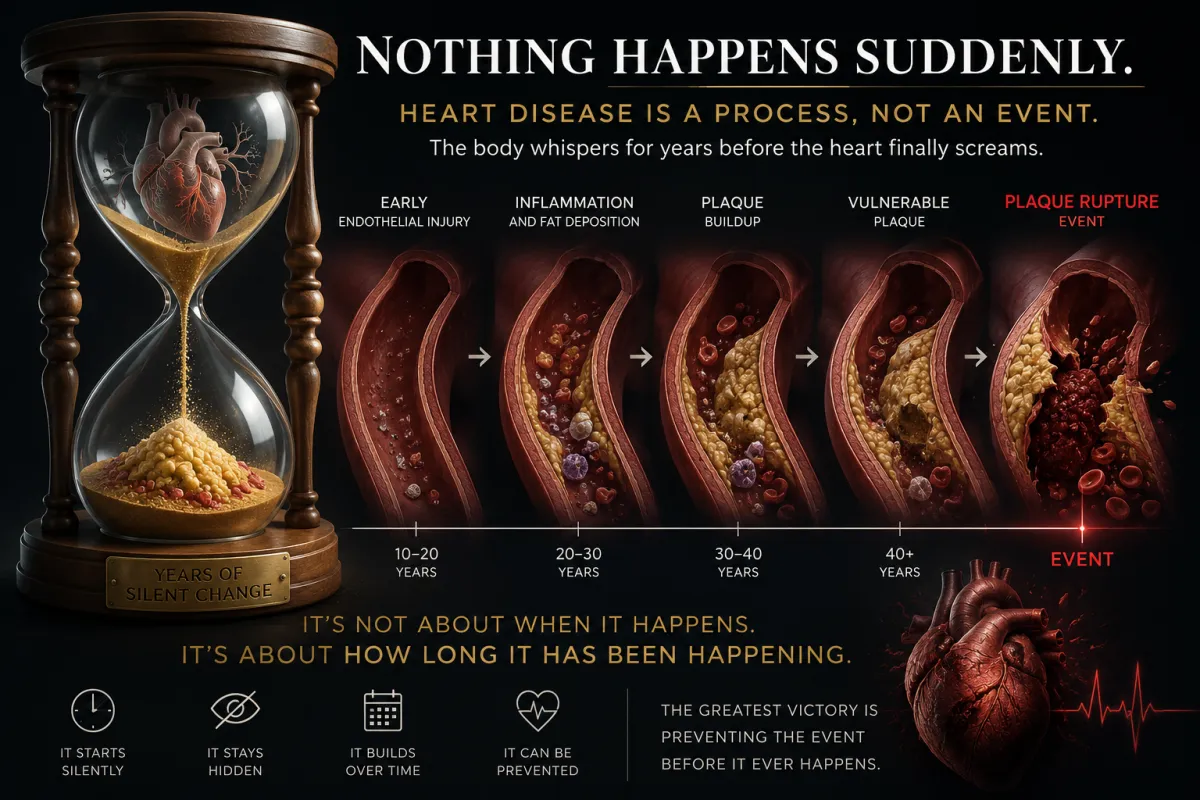
Most heart attacks are not sudden events, they are the final expression of years of silent disease
Dr John Sciales
Director, CardioCore Metabolic Wellness Center
Summary
Cardiovascular disease is still widely perceived as an abrupt and unpredictable event, despite overwhelming biological evidence demonstrating that atherosclerosis develops silently and progressively over many years before clinical manifestation. Modern cardiovascular medicine has traditionally relied heavily on statistical risk estimation through measurements such as cholesterol, blood pressure, glucose values, and population-based risk calculators, yet these markers frequently fail to identify the actual structural presence, biological behavior, and inflammatory activity of disease already developing within the arterial wall. As a result, many patients remain falsely reassured by “normal” numbers while biologically active plaque continues to evolve silently beneath the surface.
Importantly, many cardiovascular events arise not from severely obstructive lesions, but from non-obstructive, biologically vulnerable plaque that may remain entirely undetected by traditional stress testing and symptom-based evaluation. Emerging evidence from coronary artery calcium (CAC) scanning, coronary CT angiography (CCTA), inflammatory biology, plaque characterization, and cardiometabolic research increasingly supports a major paradigm shift away from viewing cardiovascular disease solely as a cholesterol-storage disorder and toward understanding it as a chronic inflammatory, immune-mediated, endothelial, thrombotic, and metabolic disease process. The artery itself carries the long biological history of disease progression long before symptoms appear, long before obstruction develops, and often long before traditional testing identifies danger.
This article reviews the biological stages of atherosclerosis, the limitations of conventional risk assessment, the importance of imaging-based structural identification of disease, and the growing need to identify upstream biological drivers such as insulin resistance, chronic inflammation, oxidative stress, endothelial dysfunction, autonomic imbalance, and prothrombotic physiology before catastrophic cardiovascular events occur. The central premise is both biologically and clinically profound: heart attacks do not happen suddenly; rather, they represent the final clinical expression of a long-standing biological process that medicine frequently recognizes far too late.
“The heart attack is not the beginning of the disease. It is the moment the disease finally becomes impossible to ignore.”
Introduction
For decades, cardiovascular disease has been described primarily through the language of risk. Patients are categorized according to cholesterol values, blood pressure measurements, diabetic status, smoking history, age, family history, and population-based risk calculators designed to estimate the probability of future cardiovascular events.[1,2] These tools have unquestionably improved public health awareness and have helped guide preventive therapy across large populations. Yet despite these advances, an important conceptual problem has quietly emerged within cardiovascular medicine: the tendency to confuse risk estimation with actual disease identification. Medicine has become exceptionally skilled at predicting who may someday develop disease, while often failing to determine whether disease is already structurally present and biologically active within the arteries themselves.
This distinction is critically important because cardiovascular disease is fundamentally structural and biological, not merely statistical. A patient may present with relatively “normal” laboratory values while simultaneously harboring extensive biologically active plaque throughout the coronary circulation, while another individual with elevated risk markers may demonstrate comparatively limited structural disease burden.[3] The artery itself—not the spreadsheet, calculator, or probability model—ultimately determines cardiovascular outcomes. The biological behavior of plaque, including its inflammatory activity, composition, vulnerability, and thrombogenic potential, frequently matters more than isolated numerical values viewed outside the context of the actual arterial environment.
The widespread perception that heart attacks occur suddenly further reinforces this misunderstanding. Families frequently describe myocardial infarction, stroke, arrhythmia, or sudden cardiac death as abrupt and unexpected events arising without warning, as though the disease process appeared instantaneously. Biologically, however, atherosclerosis is neither sudden nor random. It represents a prolonged continuum involving endothelial injury, chronic inflammation, immune recruitment, oxidative stress, lipid accumulation, plaque maturation, calcification, and eventual plaque destabilization and rupture.[4,5] By the time a cardiovascular event becomes clinically visible, the underlying biology has often been evolving silently for years or decades beneath the surface. The “sudden” event is therefore not truly the beginning of disease, but rather the final clinical expression of a process that has remained biologically active and structurally progressive long before symptoms emerged.
Modern cardiovascular imaging has increasingly challenged traditional approaches centered primarily around symptom recognition and risk-factor estimation. Coronary artery calcium (CAC) scoring and coronary CT angiography (CCTA) now allow direct visualization of plaque burden, plaque composition, plaque phenotype, and plaque vulnerability, fundamentally shifting cardiovascular medicine from probabilistic estimation toward direct structural identification of disease itself.[6,7] These technologies allow clinicians not only to estimate risk, but to directly observe the biological footprint of disease within the artery wall. Simultaneously, contemporary inflammatory and cardiometabolic research has demonstrated that cardiovascular disease cannot be adequately understood as simply a cholesterol-storage disorder, but rather as a complex inflammatory, metabolic, immune-mediated, endothelial, and prothrombotic disease process involving the interaction of vascular biology, insulin resistance, oxidative stress, chronic inflammation, thrombosis, autonomic imbalance, and metabolic dysfunction.[8-10]
This evolving understanding carries profound implications for prevention. If cardiovascular disease represents a long-standing inflammatory and cardiometabolic process rather than an abrupt event, then waiting for symptoms or advanced obstruction may represent intervention far too late in the biological timeline. The future of cardiovascular prevention increasingly depends upon earlier recognition of disease activity, earlier identification of vulnerable biology, and earlier intervention directed toward the upstream drivers responsible for plaque formation, progression, destabilization, and thrombosis before catastrophic clinical events occur.
This article explores the concept that “nothing happens suddenly” in cardiovascular disease and argues for a fundamental paradigm shift toward earlier biological recognition, imaging-guided structural identification of disease, upstream cardiometabolic prevention, and a deeper understanding of the inflammatory and metabolic drivers responsible for plaque development and destabilization long before the heart attack, stroke, or sudden cardiac event finally occurs.
“We have spent decades estimating the probability of disease while too often failing to look directly at the disease itself.”
Cardiovascular Disease: Event Versus Timeline
One of the greatest misconceptions in cardiovascular medicine is the belief that cardiovascular disease begins at the moment symptoms appear. Chest pain, dyspnea, myocardial infarction, arrhythmia, heart failure, and sudden cardiac death are commonly perceived as the starting point of disease rather than what they truly represent: the late clinical manifestations of an already advanced and biologically active process that has often been developing silently for years or decades.[4] This misunderstanding has profoundly shaped the culture of modern cardiovascular medicine by reinforcing a reactive model of care in which intervention frequently occurs only after symptoms emerge, blood flow becomes impaired, or a catastrophic event finally forces recognition of disease.
In reality, cardiovascular disease is far better understood as a timeline rather than an isolated event. The so-called “sudden” heart attack is rarely sudden biologically. Instead, it often represents the final clinical expression of progressive endothelial injury, chronic inflammation, immune activation, oxidative stress, lipid accumulation, plaque maturation, calcification, and eventual plaque destabilization occurring silently within the artery wall over many years.[5] Long before the patient experiences chest discomfort, shortness of breath, or an abnormal stress test, the biology of disease has frequently already been active beneath the surface. The artery has often been undergoing structural and inflammatory change long before medicine or the patient recognizes that danger exists.
This misunderstanding carries major clinical consequences because it encourages medicine to focus downstream on symptoms and obstruction rather than upstream on biology and disease progression. Patients and clinicians alike often wait for ischemia, angina, flow limitation, arrhythmia, or crisis before aggressive evaluation or intervention occurs, despite the fact that plaque development and vascular injury have usually been progressing silently long beforehand. By the time symptoms appear, the disease process is frequently no longer early but relatively advanced, meaning the biological timeline began many years before the clinical timeline became visible.
The concept that cardiovascular disease develops silently long before overt manifestation is strongly supported by pathological and imaging data. Autopsy studies have demonstrated early atherosclerotic lesions and fatty streak formation in adolescents and young adults, even in the absence of symptoms or recognized cardiovascular disease.[11] Similarly, prospective imaging studies utilizing coronary artery calcium (CAC) scoring and coronary CT angiography (CCTA) have repeatedly demonstrated that plaque burden commonly exists years before detectable ischemia, flow limitation, or cardiovascular events emerge.[6,7] These findings reinforce an uncomfortable but biologically important reality: absence of symptoms does not equal absence of disease.
This disconnect between biological disease progression and clinical symptom development helps explain why so many cardiovascular events are perceived as “unexpected.” In many cases, the disease itself was not unexpected biologically; rather, it remained unidentified because medicine often focuses more heavily on estimating future probability than directly visualizing present disease. The patient who “suddenly” suffers a myocardial infarction may have been carrying vulnerable plaque, endothelial dysfunction, chronic inflammation, and active cardiometabolic disease silently for decades before the event finally occurred.
Ultimately, cardiovascular disease does not begin in the emergency room, the catheterization laboratory, or at the moment chest pain develops. It begins much earlier, quietly unfolding within the biology of the artery wall while patients continue to feel entirely normal. Recognizing cardiovascular disease as a timeline rather than an event fundamentally changes the philosophy of prevention because it shifts the focus away from reacting to crisis and toward identifying disease while the biology is still silent, active, and potentially modifiable.
“The heart attack may appear sudden clinically, but biologically it is often the final chapter of a story the artery has been writing for decades.”
Risk Estimation Versus Structural Disease Identification
Traditional cardiovascular prevention has largely been organized around risk estimation models derived from epidemiologic observations and population-level associations.[1,2] Cholesterol measurements, blood pressure values, diabetic status, smoking history, obesity, age, sex, and family history remain critically important components of cardiovascular assessment and unquestionably contribute valuable information regarding future cardiovascular probability. However, these markers primarily estimate the likelihood that disease may exist or eventually develop; they do not directly identify whether biologically active atherosclerotic disease is already structurally present within the arteries themselves.
This distinction is clinically profound because population-based statistical models do not necessarily define the biology occurring inside an individual patient’s artery wall.[3] A patient may present with “normal” LDL cholesterol, normal glucose values, and acceptable blood pressure while simultaneously harboring extensive non-obstructive and biologically vulnerable plaque throughout the coronary circulation. Conversely, another patient with elevated LDL cholesterol or multiple traditional risk factors may demonstrate comparatively limited structural plaque burden and relatively stable vascular biology.[12] The discrepancy exists because cardiovascular disease is ultimately governed not simply by isolated laboratory values, but by the interaction between inflammation, endothelial function, immune activation, thrombogenicity, oxidative stress, insulin resistance, vascular remodeling, and plaque biology occurring directly within the arterial environment.
Risk calculators further illustrate this limitation because they estimate statistical probability based upon population trends rather than direct visualization of disease itself. Younger patients with significant inflammatory burden, insulin resistance, hyperinsulinemia, visceral adiposity, or aggressive cardiometabolic dysfunction may be categorized as “low risk” despite already carrying active plaque burden that has not yet become clinically apparent. In contrast, older individuals may appear “high risk” mathematically because of age alone despite demonstrating relatively stable plaque biology and minimal active disease progression. The artery itself frequently tells a far more accurate story than the statistical model designed to predict it.
Modern cardiovascular imaging has fundamentally challenged this traditional paradigm by allowing clinicians to directly visualize structural disease rather than merely estimate its probability. Coronary artery calcium (CAC) scoring and coronary CT angiography (CCTA) represent a major shift in cardiovascular medicine because they move assessment away from theoretical risk and toward direct anatomical identification of disease itself.[6,7] CAC scoring identifies calcified plaque within the coronary arteries and provides objective evidence that atherosclerosis is structurally present, while CCTA allows visualization of plaque burden, plaque morphology, non-calcified plaque, low-attenuation plaque, positive remodeling, and other characteristics associated with biologically vulnerable lesions.[7,13] For the first time, clinicians are increasingly able not only to estimate risk, but to directly observe the biological and structural footprint of disease within the coronary circulation.
This transition from risk estimation to disease identification is extraordinarily important because plaque biology often matters more than obstruction severity alone. Many cardiovascular events arise from non-obstructive yet biologically active plaque that may remain invisible to traditional symptom-based evaluation and stress testing.[13] The patient who experiences a myocardial infarction may never have developed severe enough luminal narrowing to produce ischemia during exercise testing, yet may still harbor vulnerable inflammatory plaque capable of rupture and thrombosis. Imaging therefore reveals a reality that traditional risk-factor analysis frequently misses: disease can be biologically dangerous long before it becomes obstructive.
Once plaque is visualized, one fact becomes immediately undeniable: the disease has existed for a long time. Calcification is not the beginning of atherosclerosis, but rather evidence of chronic vascular injury and long-standing plaque evolution.[6] Calcium deposition represents the body’s attempt to stabilize inflamed plaque over time, meaning that a positive CAC score is effectively a biological record of years or decades of prior vascular injury. Similarly, the presence of mixed plaque, non-calcified plaque, or high-risk plaque characteristics on CCTA reflects a prolonged inflammatory and metabolic process that has often remained silent long before clinical symptoms appeared.
This understanding fundamentally changes the philosophy of prevention because it reveals that cardiovascular disease is frequently already present before medicine formally recognizes it. The question therefore shifts from “What is this patient’s future risk?” to a far more biologically meaningful question: “Is disease already present, and if so, what is its biological behavior?” This distinction moves cardiovascular medicine away from passive observation and toward earlier identification of active disease before plaque destabilization, thrombosis, myocardial infarction, or sudden cardiac death finally occur.
“Risk calculators estimate probability, but the artery reveals reality.”
The Biology of Atherosclerosis
Atherosclerosis is increasingly recognized not as a passive process of cholesterol accumulation, but as a chronic inflammatory, immune-mediated, endothelial, thrombotic, and metabolic disease process involving highly active vascular biology.[4,8,9] For many years, cardiovascular disease was simplified into the concept of cholesterol gradually “clogging” arteries in a manner similar to debris accumulating within a pipe. Although lipoproteins clearly play a central role in plaque formation, contemporary research has demonstrated that the biology of atherosclerosis is vastly more complex and involves the interaction of endothelial dysfunction, chronic inflammation, immune activation, oxidative stress, thrombogenic signaling, metabolic dysfunction, and vascular injury occurring continuously within the artery wall.[4,5]
The process begins with endothelial injury and dysfunction, which represents one of the earliest and most important biological events in cardiovascular disease progression.[14-16] The endothelium is a thin single-cell layer lining the inner surface of blood vessels and serves as a highly active biological interface regulating vascular tone, nitric oxide signaling, thrombosis, permeability, inflammation, immune interaction, and blood flow dynamics. Under normal conditions, the endothelium functions as a protective barrier maintaining vascular stability and anti-inflammatory balance. However, chronic exposure to hypertension, insulin resistance, hyperglycemia, smoking, oxidative stress, abnormal sleep, sedentary behavior, sympathetic nervous system overactivation, environmental toxins, visceral adiposity, and chronic inflammatory physiology gradually injures this protective lining and alters its biological behavior.[14-16]
Once endothelial dysfunction develops, the artery begins to shift from a protected anti-inflammatory environment toward a biologically vulnerable and pro-inflammatory state. Nitric oxide production becomes impaired, vascular permeability increases, oxidative stress intensifies, and inflammatory signaling pathways become activated.[4,5] Adhesion molecules are expressed along the endothelial surface, effectively transforming the artery wall into a site of immune recruitment and inflammatory activity. At this stage, the disease process is already biologically active despite the complete absence of symptoms, which explains why atherosclerosis can silently progress for many years before clinical recognition occurs.
As inflammatory signaling intensifies, circulating monocytes migrate into the arterial intima and differentiate into macrophages, which begin ingesting oxidized LDL particles trapped within the vessel wall.[4] These macrophages gradually become engorged with lipid material and transform into foam cells, producing the fatty streaks that represent the earliest visible manifestations of atherosclerosis. Importantly, this process is not passive lipid storage but active vascular immunology involving cytokine signaling, inflammatory amplification, oxidative modification of lipoproteins, endothelial activation, and immune-cell recruitment.[4,8] The artery effectively becomes the site of a chronic inflammatory process that continuously perpetuates its own progression.
Over time, persistent inflammation, oxidative stress, endothelial injury, and immune activation allow these fatty streaks to evolve into mature atherosclerotic plaque containing lipid-rich necrotic cores, inflammatory cells, fibrous tissue, smooth muscle proliferation, calcification, and areas of biological instability.[5] As plaque progresses, the vessel wall undergoes remodeling, and calcium deposition frequently develops as part of the body’s attempt to stabilize chronically inflamed lesions. However, even plaques that appear relatively stable structurally may still harbor significant inflammatory activity beneath the surface.
Perhaps most importantly, modern cardiovascular biology has demonstrated that plaque vulnerability depends not simply upon plaque size or degree of obstruction, but upon plaque composition and biological behavior.[5,17] Plaques rich in inflammatory activity, macrophage infiltration, lipid content, oxidative stress, necrotic debris, and thin fibrous caps may carry substantial rupture risk despite relatively modest luminal narrowing. This explains why many myocardial infarctions arise from non-obstructive lesions that may not significantly impair blood flow prior to rupture.[17] The danger therefore lies not merely in how narrow the artery becomes, but in how biologically unstable the plaque itself has become.
This understanding fundamentally changes the conceptual framework of cardiovascular disease. The plaque is not simply an inert blockage slowly restricting blood flow like rust accumulating within a pipe. Rather, it is a biologically active inflammatory lesion interacting continuously with immune signaling, endothelial dysfunction, thrombosis, oxidative stress, and metabolic physiology. The artery wall becomes an active battlefield of inflammation, vascular injury, immune activation, and attempted repair long before symptoms appear or obstruction becomes clinically evident.
Recognizing atherosclerosis as a dynamic inflammatory and cardiometabolic disease process carries profound implications for prevention and treatment. It suggests that meaningful cardiovascular prevention cannot focus exclusively on lowering isolated laboratory numbers, but must also address the upstream biology driving endothelial injury, inflammation, oxidative stress, immune activation, insulin resistance, and vascular instability before plaque rupture and thrombosis finally occur.
“Atherosclerosis is not simply cholesterol stored in the artery wall; it is the biological scar of years of inflammation, metabolic dysfunction, and vascular injury.”
Why Stress Testing Frequently Misses Dangerous Disease
Traditional cardiac stress testing has played an important role in cardiovascular medicine for decades and remains useful for evaluating ischemia, exercise tolerance, symptom reproduction, and advanced obstructive coronary disease.[18] However, one of the most important misunderstandings in modern cardiology is the assumption that a “normal” stress test means the absence of significant cardiovascular disease. In reality, stress testing evaluates a very specific physiological question: whether blood flow to the heart muscle becomes significantly impaired during exertion because of severe luminal obstruction. It does not directly evaluate the presence of atherosclerosis, plaque burden, plaque biology, endothelial dysfunction, inflammation, plaque vulnerability, or the underlying cardiometabolic environment driving disease progression.
This distinction is critically important because stress testing is fundamentally a downstream ischemia test rather than an upstream disease-identification test. The test becomes abnormal primarily when an artery has narrowed sufficiently—often approaching 70% luminal obstruction or greater—to impair coronary blood flow during stress or exertion.[18] Yet most myocardial infarctions do not arise from these severely obstructive, stable lesions. Instead, the majority of acute coronary syndromes originate from biologically vulnerable plaques that may produce less than 50% luminal narrowing prior to rupture.[17,19] These plaques are frequently rich in inflammatory activity, lipid content, macrophage infiltration, oxidative stress, and thin fibrous caps, making them biologically unstable despite appearing only mildly obstructive anatomically.
Consequently, a patient may harbor extensive inflammatory plaque burden throughout the coronary circulation while still demonstrating a “normal” stress test because blood flow has not yet become sufficiently impaired to produce detectable ischemia during exercise.[18] This explains why many patients experience myocardial infarction, sudden cardiac death, or acute coronary syndromes shortly after reassuring stress test results. The stress test may correctly identify the absence of severe flow limitation while simultaneously failing to identify the underlying plaque biology responsible for future rupture and thrombosis. In this sense, the test is functioning exactly as designed, but the biology of disease extends far beyond what the test was built to evaluate.
This limitation reflects a much larger biological reality within cardiovascular disease: ischemia and obstruction are often relatively late manifestations of atherosclerosis. Endothelial dysfunction, chronic inflammation, oxidative stress, immune activation, insulin resistance, plaque formation, vascular remodeling, and thrombogenic physiology frequently develop years or decades before sufficient luminal narrowing occurs to impair blood flow during stress testing.[4,5,14-16] By the time a lesion becomes severely obstructive enough to produce ischemia, the disease process itself is often already advanced.
Importantly, stress testing does not tell clinicians whether a patient actually has atherosclerosis. It does not directly identify plaque burden. It does not evaluate non-calcified plaque, low-attenuation plaque, vulnerable plaque, or inflammatory plaque activity. It does not characterize plaque composition, vascular inflammation, endothelial injury, or the biological instability of the arterial wall. In many ways, stress testing evaluates the physiological consequence of advanced disease rather than the disease process itself.
This distinction helps explain why decades of treating stable obstructive disease based primarily upon ischemia have not consistently demonstrated major reductions in long-term mortality in many patient populations when compared with intensive medical therapy alone.[18] Procedures designed to relieve obstruction and improve symptoms, such as percutaneous coronary intervention (PCI), may significantly improve angina and quality of life in selected patients, but they do not necessarily eliminate the underlying inflammatory and cardiometabolic biology driving plaque progression throughout the remainder of the coronary tree. Treating a focal obstruction does not automatically stabilize vulnerable plaque elsewhere, nor does it correct endothelial dysfunction, chronic inflammation, insulin resistance, oxidative stress, autonomic imbalance, or prothrombotic physiology that continue driving disease progression beneath the surface.
This is why many patients continue to experience cardiovascular events despite previous stenting or apparently successful revascularization procedures. The visible obstruction may have been treated, yet the underlying vascular biology often remains active. The true disease process extends far beyond a single narrowed segment within the artery.
Modern coronary imaging has increasingly highlighted these limitations. Coronary artery calcium (CAC) scanning and coronary CT angiography (CCTA) allow clinicians to directly visualize plaque burden, plaque composition, and plaque vulnerability rather than simply evaluating downstream ischemia.[6,7] CCTA can identify non-obstructive plaque, positive remodeling, low-attenuation plaque, and other high-risk characteristics associated with vulnerable lesions long before severe luminal obstruction develops.[7,13] This fundamentally shifts cardiovascular evaluation away from asking only whether flow is impaired and toward asking whether biologically dangerous disease is already present.
The discussion surrounding radiation exposure further reinforces the need to reconsider traditional assumptions about cardiovascular testing. Many nuclear stress tests expose patients to radiation doses ranging approximately between 9 and 14 millisieverts (mSv), depending upon the protocol utilized, while modern CAC scoring and contemporary CCTA protocols frequently involve substantially lower radiation exposure, often in the range of approximately 1 to 1.5 mSv.[6,7] For perspective, annual natural background radiation exposure averages roughly 3 mSv, while a mammogram delivers approximately 0.7 mSv. In other words, medicine has often accepted relatively high-radiation downstream ischemia testing designed to identify advanced obstructive disease while historically underutilizing lower-radiation imaging modalities capable of identifying earlier structural disease and vulnerable plaque biology.
This evolving understanding forces an important philosophical shift in cardiovascular medicine. The central question should no longer simply be whether a patient has flow-limiting obstruction severe enough to produce ischemia during exertion. The more important question may be whether biologically active atherosclerotic disease is already present within the arteries long before ischemia, symptoms, or catastrophic plaque rupture occur.
Ultimately, stress testing remains valuable within the appropriate clinical context, particularly for evaluating symptoms, ischemia, exercise capacity, and advanced obstructive disease. However, it was never designed to fully evaluate the biology of atherosclerosis itself. Cardiovascular disease is not merely a problem of obstruction and blood flow; it is fundamentally a chronic inflammatory, endothelial, thrombotic, metabolic, and immune-mediated disease process that often remains biologically active long before traditional stress testing becomes abnormal.
“A normal stress test does not necessarily mean a normal artery; it often means the disease has not yet become obstructive enough to reveal itself.”
Inflammation, Metabolism, and Cardiometabolic Disease
Over the past two decades, cardiovascular medicine has undergone a profound biological shift in understanding, moving away from the traditional view that atherosclerosis is simply a cholesterol-storage disorder and toward recognizing cardiovascular disease as a chronic inflammatory, immune-mediated, endothelial, thrombotic, and metabolic process.[8,9] While cholesterol remains critically important in plaque formation and progression, modern research has increasingly demonstrated that lipoproteins alone do not fully explain why plaque forms, why plaque progresses, why plaque destabilizes, or why cardiovascular events continue to occur even in patients with aggressively lowered LDL cholesterol levels. The biology underlying cardiovascular disease is far more complex and involves the interaction between inflammation, endothelial dysfunction, immune signaling, oxidative stress, thrombosis, autonomic imbalance, insulin resistance, and metabolic dysfunction occurring continuously within the vascular environment.
One of the most important pieces of evidence supporting the inflammatory nature of cardiovascular disease emerged from the landmark CANTOS trial, which demonstrated that targeting inflammation with canakinumab reduced cardiovascular events independently of major LDL cholesterol reduction.[10] This finding was biologically profound because it demonstrated that cardiovascular risk could be modified through direct suppression of inflammatory signaling even without substantially altering cholesterol levels themselves. Furthermore, reductions in cardiovascular events correlated closely with reductions in inflammatory markers such as high-sensitivity C-reactive protein (hsCRP), reinforcing the concept that inflammation is not merely associated with cardiovascular disease but actively participates in its progression.[10]
This inflammatory framework helps explain many observations that traditional cholesterol-centered models struggle to fully account for. Patients with relatively normal cholesterol levels may still experience premature cardiovascular disease, while others with elevated cholesterol may remain free of clinical events for many years. The difference often lies not simply in cholesterol concentration, but in the broader biological environment in which vascular injury, inflammation, oxidative stress, endothelial dysfunction, and immune activation are occurring. Plaque vulnerability is frequently driven not only by the amount of plaque present, but by the inflammatory activity within the plaque itself.
At the center of this evolving understanding is the growing recognition that insulin resistance and metabolic dysfunction play a major upstream role in cardiovascular disease development.[14-16,20] Insulin resistance is not merely a glucose disorder; it represents a systemic metabolic and vascular condition that affects endothelial biology, inflammatory signaling, oxidative stress, thrombosis, vascular tone, lipid metabolism, autonomic balance, and immune activity throughout the body. Long before fasting glucose or hemoglobin A1c become abnormal, hyperinsulinemia and impaired metabolic flexibility may already be exerting substantial biological effects on the vascular system.
Under normal physiological conditions, insulin supports endothelial nitric oxide production, vascular relaxation, glucose uptake, and metabolic homeostasis. However, as insulin resistance develops, these protective pathways become impaired while pro-inflammatory and pro-atherogenic signaling pathways become amplified.[14] The vascular environment gradually shifts toward endothelial dysfunction, oxidative stress, inflammatory cytokine production, sympathetic nervous system overactivation, prothrombotic physiology, and vascular remodeling. The artery becomes biologically vulnerable long before overt diabetes is formally diagnosed.
This has enormous implications because many patients currently considered metabolically “normal” based upon fasting glucose or hemoglobin A1c may already demonstrate significant cardiometabolic dysfunction.[20] Standard laboratory thresholds often identify disease relatively late in the biological timeline, whereas insulin resistance and hyperinsulinemia may remain active silently for many years beforehand. During this period, vascular injury, inflammation, plaque formation, visceral adiposity, endothelial dysfunction, and oxidative stress may continue progressing despite apparently “normal” routine laboratory testing.
Visceral adiposity itself further amplifies this inflammatory and metabolic process. Adipose tissue, particularly visceral fat surrounding abdominal organs, is now understood to function as a biologically active endocrine and inflammatory organ rather than merely an inert storage site for excess calories.[15,16] Visceral adipose tissue releases inflammatory cytokines, free fatty acids, adipokines, and hormonal signals that contribute directly to insulin resistance, endothelial dysfunction, vascular inflammation, oxidative stress, and thrombogenic physiology. This helps explain why body composition and metabolic health often carry greater biological importance than body weight alone.
Chronic sympathetic nervous system activation also appears increasingly intertwined with cardiometabolic disease progression.[16] Persistent stress physiology, abnormal sleep, autonomic imbalance, poor recovery, and chronic elevations in catecholamine signaling may contribute to endothelial dysfunction, inflammation, hypertension, insulin resistance, oxidative stress, vascular stiffness, and adverse metabolic remodeling. Cardiovascular disease therefore cannot be adequately understood solely through isolated laboratory measurements, because the biology driving disease frequently involves multiple interconnected systems operating simultaneously beneath the surface.
Importantly, this inflammatory and metabolic framework also helps explain the persistent residual cardiovascular risk observed even after aggressive LDL reduction.[8-10] While statin therapy and LDL lowering clearly reduce cardiovascular events and remain foundational components of prevention, many patients continue to experience myocardial infarction, stroke, plaque progression, and cardiovascular mortality despite achieving acceptable cholesterol targets. This residual risk likely reflects the continued presence of inflammatory biology, insulin resistance, endothelial dysfunction, oxidative stress, thrombogenic signaling, and metabolic disease that remain active despite improved lipid values.
The implications of this evolving understanding are profound because they fundamentally redefine cardiovascular disease itself. Atherosclerosis is no longer adequately described as simply a disorder of cholesterol accumulation within the artery wall. It is increasingly recognized as an immune-inflammatory-metabolic disease involving endothelial injury, oxidative stress, thrombosis, vascular inflammation, autonomic imbalance, metabolic dysfunction, and chronic biological instability occurring throughout the vascular system.[8-10] The plaque therefore becomes not merely a passive obstruction but a visible structural expression of deeper systemic biological dysfunction.
This shift also fundamentally changes the philosophy of prevention. If cardiovascular disease is driven by chronic inflammatory and metabolic biology long before symptoms occur, then meaningful prevention must move upstream toward earlier recognition and modification of these underlying drivers. Waiting until glucose becomes frankly diabetic, until obstruction becomes flow-limiting, or until symptoms appear may represent intervention far too late within the biological timeline of disease progression.
Ultimately, the future of cardiovascular medicine will increasingly depend not simply upon lowering numbers, but upon understanding and altering the biological terrain that allows vascular injury, inflammation, thrombosis, endothelial dysfunction, and plaque instability to develop in the first place.
“The plaque inside the artery is often only the visible scar of a much deeper inflammatory and metabolic disease already occurring throughout the body.”
The Fire Beneath the Smoke
One of the greatest limitations in modern chronic disease management is the tendency to focus predominantly on the visible downstream manifestations of disease while failing to adequately identify and address the deeper upstream biology driving those manifestations in the first place. Elevated blood pressure, hyperglycemia, dyslipidemia, atrial fibrillation, obesity, fatty liver disease, vascular calcification, heart failure, and cardiovascular events are frequently approached as separate isolated disorders requiring separate isolated treatments, despite the fact that they are often interconnected expressions of the same underlying cardiometabolic and inflammatory biology. The visible abnormality becomes the focus of treatment, while the biological environment responsible for creating that abnormality frequently remains active beneath the surface.
This distinction can be understood through the analogy of smoke and fire. In many patients, elevated glucose is smoke. Hypertension is smoke. Elevated triglycerides are smoke. Low HDL cholesterol is smoke. Visceral obesity is smoke. Fatigue, endothelial dysfunction, vascular stiffness, atrial fibrillation, fatty liver disease, and eventually plaque formation are all smoke. The deeper biological “fire” frequently involves insulin resistance, chronic inflammation, oxidative stress, endothelial dysfunction, autonomic imbalance, mitochondrial dysfunction, chronic sympathetic nervous system activation, sleep disruption, visceral adiposity, metabolic inflexibility, immune activation, and prothrombotic physiology occurring silently long before catastrophic cardiovascular disease develops.[14-16]
The problem with treating only smoke is that the underlying fire frequently continues burning despite apparent improvement in downstream markers. A patient’s blood pressure may improve numerically while endothelial dysfunction and inflammatory signaling remain active. Glucose levels may normalize while hyperinsulinemia and insulin resistance persist beneath the surface. LDL cholesterol may decline while oxidative stress, plaque inflammation, thrombogenic biology, visceral adiposity, and vascular instability continue progressing. The visible marker improves, yet the upstream disease-driving biology often remains incompletely addressed. This helps explain why many patients continue progressing toward cardiovascular disease despite apparently “controlled” laboratory values.
This concept becomes particularly important in cardiovascular disease because plaque progression and plaque destabilization are fundamentally biological processes rather than simply numerical abnormalities.[4,8,9] The artery does not respond solely to cholesterol concentration or blood pressure values in isolation. Rather, the vascular environment responds to the total biological terrain surrounding it, including inflammatory signaling, oxidative stress, endothelial injury, autonomic balance, thrombogenicity, metabolic dysfunction, insulin signaling, hormonal physiology, sleep quality, environmental stressors, body composition, mitochondrial energy regulation, and chronic immune activation. When this biological terrain remains chronically abnormal, the artery continues to experience injury and inflammatory activation even when isolated downstream laboratory markers appear “controlled.”
Insulin resistance represents one of the clearest examples of this upstream biology.[14,15] Long before fasting glucose becomes frankly diabetic, insulin resistance may already be promoting endothelial dysfunction, inflammatory cytokine production, oxidative stress, vascular remodeling, sympathetic nervous system overactivation, thrombosis, visceral fat accumulation, and metabolic inflexibility. During this prolonged preclinical period, patients are frequently reassured by “normal” glucose values despite the fact that vascular injury may already be actively progressing beneath the surface. The biology of disease therefore often begins years or even decades before traditional laboratory thresholds finally become abnormal enough to establish a formal diagnosis.
Similarly, chronic inflammation functions as a major upstream driver connecting multiple downstream disease manifestations simultaneously.[8-10] Inflammatory signaling contributes directly to endothelial dysfunction, plaque instability, insulin resistance, thrombosis, atrial remodeling, vascular stiffness, hypertension, heart failure, and accelerated atherosclerosis. Yet inflammation itself often remains relatively invisible during routine clinical evaluation unless specifically sought and biologically understood. This helps explain why cardiovascular disease frequently continues progressing despite treatment strategies directed exclusively toward isolated downstream markers rather than the inflammatory and metabolic biology driving disease progression itself.
Autonomic imbalance and chronic sympathetic nervous system activation further amplify this process.[16] Persistent elevations in stress physiology, catecholamine signaling, poor sleep, impaired recovery, emotional stress, sedentary behavior, and metabolic dysfunction collectively contribute to endothelial injury, hypertension, insulin resistance, oxidative stress, inflammatory amplification, vascular stiffness, arrhythmogenesis, and thrombogenicity. Cardiovascular disease therefore frequently reflects not a single isolated abnormal pathway, but rather the convergence of multiple chronic biological stressors operating simultaneously over long periods of time.
This framework fundamentally changes how prevention should be approached. The future of cardiovascular medicine cannot rely exclusively on suppressing visible manifestations of disease after they appear. Lowering blood pressure without understanding why vascular dysfunction developed, lowering glucose without evaluating insulin resistance, or lowering LDL cholesterol without assessing inflammatory biology may improve selected downstream markers while leaving the underlying disease process incompletely addressed. True prevention requires understanding not only which number became abnormal, but why the biological environment became unstable enough to produce disease in the first place.
This is where the concept of cardiometabolic medicine becomes increasingly important. Cardiovascular disease does not develop independently from metabolism, inflammation, sleep, autonomic balance, body composition, hormonal signaling, mitochondrial health, environmental exposure, nutrition, and immune regulation. The artery reflects the cumulative biological environment surrounding it. Plaque itself often becomes the structural footprint of years of inflammatory and metabolic dysfunction occurring throughout the body.
Ultimately, if medicine focuses only on smoke, the fire beneath it may continue burning silently for years until it finally manifests as myocardial infarction, stroke, arrhythmia, heart failure, renal disease, or sudden cardiac death. The future of prevention therefore depends upon identifying and extinguishing the upstream biological fire before the downstream smoke becomes clinically catastrophic.
“Blood pressure, glucose, cholesterol, and plaque are often not the fire itself—they are the smoke rising from a deeper biological process already burning underneath.”
Toward a New Model of Prevention
Modern cardiovascular prevention must increasingly evolve from reactive medicine toward proactive biological recognition. Symptoms, obstruction, ischemia, arrhythmia, myocardial infarction, heart failure, and sudden cardiac death are often relatively late manifestations of disease progression rather than the beginning of disease itself.[18] By the time chest pain develops, a stress test becomes abnormal, or a cardiovascular event occurs, the underlying biology has frequently been active for years or decades. Endothelial dysfunction, inflammation, insulin resistance, oxidative stress, vascular injury, autonomic imbalance, and plaque formation usually begin long before symptoms emerge, which means the traditional strategy of waiting for clinical manifestation frequently identifies disease far too late within the biological timeline.
This evolving understanding is fundamentally reshaping the future of prevention. Advanced imaging combined with deeper metabolic, inflammatory, hormonal, and cardiometabolic evaluation now allows clinicians to identify disease while it is still biologically active and potentially modifiable, rather than waiting until irreversible structural injury and catastrophic cardiovascular events occur.[6,7] Coronary artery calcium (CAC) scoring and coronary CT angiography (CCTA) allow direct visualization of plaque burden and plaque biology, while modern cardiometabolic evaluation increasingly focuses on identifying the upstream biological drivers responsible for vascular injury long before obstruction develops.
At CardioCore Metabolic Wellness Center, this philosophy forms the foundation of how prevention is approached. The goal is not simply to manage numbers after they become abnormal, but to identify and address the deeper biology driving disease in the first place. Rather than focusing exclusively on isolated downstream markers such as cholesterol, blood pressure, or glucose, the emphasis shifts toward understanding why the vascular environment became biologically unstable enough to produce disease. Cardiovascular disease is viewed not as an isolated event or isolated organ problem, but as the structural expression of a much broader inflammatory and metabolic process occurring throughout the body.
This requires a far more comprehensive and personalized approach to prevention. CardioCore integrates precision imaging, metabolic medicine, functional medicine, inflammatory biology, and holistic cardiovascular evaluation to better understand each patient’s unique biological terrain. Genetics are evaluated to identify inherited predispositions influencing metabolism, detoxification, inflammation, thrombosis, vascular biology, and cardiovascular risk. Metabolic function is analyzed in detail because insulin resistance and impaired metabolic flexibility often represent some of the earliest drivers of vascular disease long before overt diabetes develops. Hormonal balance is assessed because cortisol physiology, testosterone, estrogen balance, thyroid function, and autonomic regulation all significantly influence inflammation, endothelial function, body composition, vascular tone, and cardiometabolic health.
Equally important, increasing attention is directed toward the gut microbiome and gastrointestinal biology because emerging evidence continues to demonstrate profound interactions between the gut, inflammation, immune signaling, metabolism, vascular function, and chronic disease progression. The gut is no longer viewed simply as a digestive organ, but as a major immunologic and metabolic interface capable of influencing systemic inflammation, oxidative stress, endothelial dysfunction, and cardiometabolic health. Nutritional quality, sleep physiology, stress biology, autonomic balance, mitochondrial function, body composition, environmental exposures, and lifestyle behavior are all recognized as important contributors to the biological environment surrounding the artery.
This integrated approach fundamentally changes the philosophy of prevention because the focus moves away from merely estimating future risk and toward directly identifying present disease and understanding the biology driving its progression. The goal is no longer simply to wait for symptoms, obstruction, or events before reacting. The goal is to identify plaque while it remains biologically vulnerable yet potentially stabilizable, to recognize metabolic dysfunction while it remains reversible, and to intervene before inflammation, thrombosis, endothelial dysfunction, and vascular instability ultimately culminate in myocardial infarction, stroke, arrhythmia, or heart failure.
This is why precision, prevention, personalization, and proactive medicine form the core pillars of the CardioCore philosophy. Every patient carries a unique biological story involving genetics, metabolism, inflammation, hormones, lifestyle, autonomic balance, gut biology, environmental exposure, and vascular physiology. Effective prevention therefore cannot rely upon generalized population averages alone. It requires individualized biological understanding aimed at changing disease trajectory before catastrophic outcomes occur.
Ultimately, the future of cardiovascular medicine will belong not to the systems that simply react to disease after it becomes clinically obvious, but to those capable of identifying and modifying the biology driving disease while it is still silent. At CardioCore Metabolic Wellness Center, the mission is to move beyond simply treating numbers and instead focus on understanding the biology, identifying the disease, changing the trajectory, and ultimately changing outcomes.
Because when disease is identified earlier, biology can be altered earlier. And when biology changes, outcomes can change with it.
“The future of prevention is no longer about waiting for disease to declare itself—it is about identifying the biology driving disease early enough to change the trajectory before the event ever occurs.”
Conclusion
Nothing in cardiovascular biology truly happens suddenly. Myocardial infarction, stroke, arrhythmia, heart failure, and sudden cardiac death are usually not isolated random events appearing without warning, but rather the final clinical manifestations of a long-standing biological process involving endothelial injury, chronic inflammation, immune activation, oxidative stress, insulin resistance, thrombosis, metabolic dysfunction, plaque progression, and eventual plaque destabilization occurring silently over many years.[4,5] The catastrophic event that appears “sudden” clinically is often only the moment the biology can no longer remain hidden.
Modern medicine now possesses imaging technology capable of directly visualizing disease years before symptoms occur, along with a rapidly expanding understanding of the inflammatory, metabolic, endothelial, autonomic, and cardiometabolic mechanisms driving vascular injury and plaque instability.[6-10] The challenge moving forward is therefore no longer whether early disease can be identified. The challenge is whether clinicians and patients are willing to move beyond passive observation, delayed reaction, and simple statistical risk estimation toward earlier biological recognition, upstream prevention, and proactive intervention before irreversible injury occurs.
This issue extends far beyond medicine alone because when a cardiovascular event occurs, it rarely affects only the individual patient. A heart attack does not impact one life; it changes entire families. Sudden cardiac death does not simply end a biological process; it leaves spouses, children, parents, friends, and loved ones carrying emotional scars that often last forever. The phone call in the middle of the night, the unexpected hospitalization, the loss of a parent, the disabled survivor unable to return to normal life—these are the human consequences of a disease process that frequently began silently many years earlier.
One of the greatest tragedies in cardiovascular disease is that people often wait until something catastrophic happens before deciding to change behavior, improve health, seek evaluation, address metabolic dysfunction, or take prevention seriously. Patients frequently ignore fatigue, weight gain, poor sleep, visceral obesity, stress, elevated glucose, hypertension, and declining metabolic health because the body remains relatively quiet while the disease silently progresses underneath. Human nature often delays action until fear arrives, until symptoms appear, or until tragedy forces attention. Yet by the time the event occurs, the biological process has frequently already been active for decades.
This is precisely why the concept that “nothing happens suddenly” is so important. The body whispers long before it screams. The artery tells the story long before the heart attack occurs. Endothelial dysfunction, inflammation, insulin resistance, oxidative stress, autonomic imbalance, and plaque formation all develop progressively over time while patients continue feeling relatively normal. The disease does not suddenly appear; rather, it slowly evolves beneath the surface until one day it becomes impossible to ignore.
The future of cardiovascular prevention therefore depends upon changing not only medical technology, but medical philosophy itself. Prevention cannot remain centered solely around waiting for symptoms, treating obstruction, or reacting after catastrophe occurs. It must increasingly focus on identifying disease while the biology remains active, vulnerable, and potentially reversible. It must focus on understanding the upstream drivers of disease rather than simply suppressing the downstream manifestations. Most importantly, it must focus on changing trajectories early enough to prevent the event from ever happening in the first place.
At CardioCore Metabolic Wellness Center, this philosophy represents the foundation of prevention. The goal is not simply to react to disease after the damage is done, but to identify the biology driving disease earlier, understand the unique inflammatory and metabolic terrain surrounding each patient, and intervene while meaningful change is still possible. Precision imaging, metabolic medicine, inflammatory evaluation, hormonal assessment, body composition analysis, gut biology, lifestyle evaluation, and personalized prevention are all integrated toward one central purpose: identifying disease before disease identifies the patient.
Ultimately, we cannot prevent every tragedy, but we can stop pretending that most cardiovascular events occur without warning. The biology has usually been speaking for years. The question is whether we are finally willing to listen early enough to act.
Because the greatest cardiovascular victory is not surviving the heart attack.
It is preventing the heart attack from ever happening at all.
“The tragedy of cardiovascular disease is not simply that people die suddenly—it is that the biology was often progressing silently for years while everyone waited for symptoms before finally paying attention.”
Book a Call and Find Out More... Dr John
CLICK HERE TO BOOK A DISCOVERY CALL
CLICK HERE TO JOIN OUR PRIVATE CardioCore COMMUNITY
CLICK HERE TO SPEAK TO OUR VIRTUAL ASSISTANT
About the Author
John Sciales, M.D. is a cardiometabolic educator, health strategist, and founder of CardioCore Metabolic Wellness Center, where his work focuses on prevention, education, and personalized health coaching through an integrative Functional Medicine and cardiometabolic framework.
Dr. Sciales’ philosophy is centered on a simple but powerful concept: chronic disease rarely begins suddenly, and meaningful prevention requires understanding the biology driving disease long before catastrophic events occur. His work emphasizes identifying the upstream contributors to cardiovascular and metabolic dysfunction, including insulin resistance, chronic inflammation, endothelial dysfunction, stress physiology, sleep disruption, nutritional imbalance, body composition abnormalities, hormonal dysfunction, and gut microbiome health.
Rather than replacing a patient’s treating physician, Dr. Sciales works to empower both patients and healthcare providers through deeper education and biological understanding. He helps individuals better understand whether disease has been appropriately identified, whether treatment strategies align with current evidence and established medical guidelines, and how lifestyle, metabolic, inflammatory, and behavioral factors may be contributing to long-term disease progression.
Central to his approach is the belief that optimal prevention requires both appropriate downstream medical evaluation and meaningful upstream biological intervention. Advanced cardiovascular imaging, metabolic evaluation, inflammatory assessment, and personalized lifestyle strategies are integrated to help patients understand not simply their risk, but the underlying biology influencing their health trajectory.
Dr. Sciales strongly supports collaborative care and welcomes communication with patients’ physicians when appropriate in order to enhance continuity, education, and overall quality of care. His ultimate goal is to help individuals move beyond reactive disease management toward a more proactive, informed, and personalized model of long-term cardiometabolic health and wellness.
References
1.Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk. Circulation. 2014;129:S49-S73.
2.Stone NJ, Robinson JG, Lichtenstein AH, et al. 2018 ACC/AHA guideline on the management of blood cholesterol. Circulation. 2019;139:e1082-e1143.
3.Budoff MJ, Young R, Lopez VA, et al. Progression of coronary calcium and incident coronary heart disease events. J Am Coll Cardiol. 2013;61(12):1231-1239.
4.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340(2):115-126.
5.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47(8 Suppl):C13-C18.
6.Budoff MJ, Shaw LJ, Liu ST, et al. Long-term prognosis associated with coronary calcification. J Am Coll Cardiol. 2007;49(18):1860-1870.
7.Newby DE, Adamson PD, Berry C, et al. Coronary CT angiography and 5-year risk of myocardial infarction. N Engl J Med. 2018;379(10):924-933.
8.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685-1695.
9.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-325.
10.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119-1131.
11.Berenson GS, Srinivasan SR, Bao W, et al. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. N Engl J Med. 1998;338:1650-1656.
12.Nasir K, Budoff MJ, Wong ND, et al. Family history of premature coronary heart disease and coronary artery calcification. Circulation. 2007;116:619-626.
13.Motoyama S, Sarai M, Harigaya H, et al. Computed tomographic angiography characteristics of atherosclerotic plaques subsequently resulting in acute coronary syndrome. J Am Coll Cardiol. 2009;54(1):49-57.
14.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595-1607.
15.Laakso M, Kuusisto J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat Rev Endocrinol. 2014;10:293-302.
16.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045-2051.
17.Naghavi M, Libby P, Falk E, et al. From vulnerable plaque to vulnerable patient. Circulation. 2003;108:1664-1672.
18.Boden WE, O’Rourke RA, Teo KK, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. 2007;356:1503-1516.
19.PROSPECT Investigators. Natural history of coronary atherosclerosis. N Engl J Med. 2011;364:226-235.
20.Jesus JM, et al. Prevalence of abnormal glucose metabolism in coronary artery disease patients with normal fasting glucose. Am J Cardiol. 2013;111:364-368.
